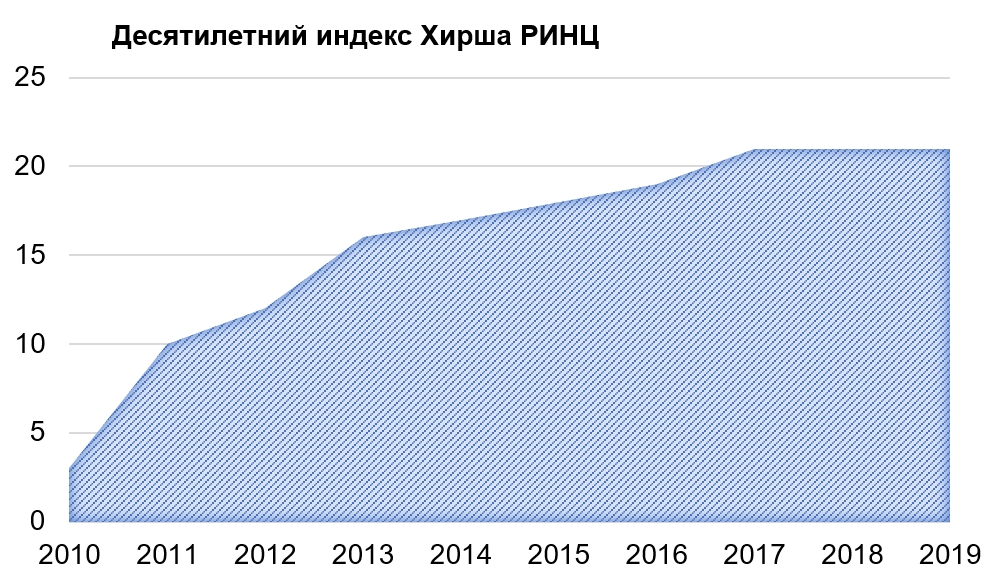
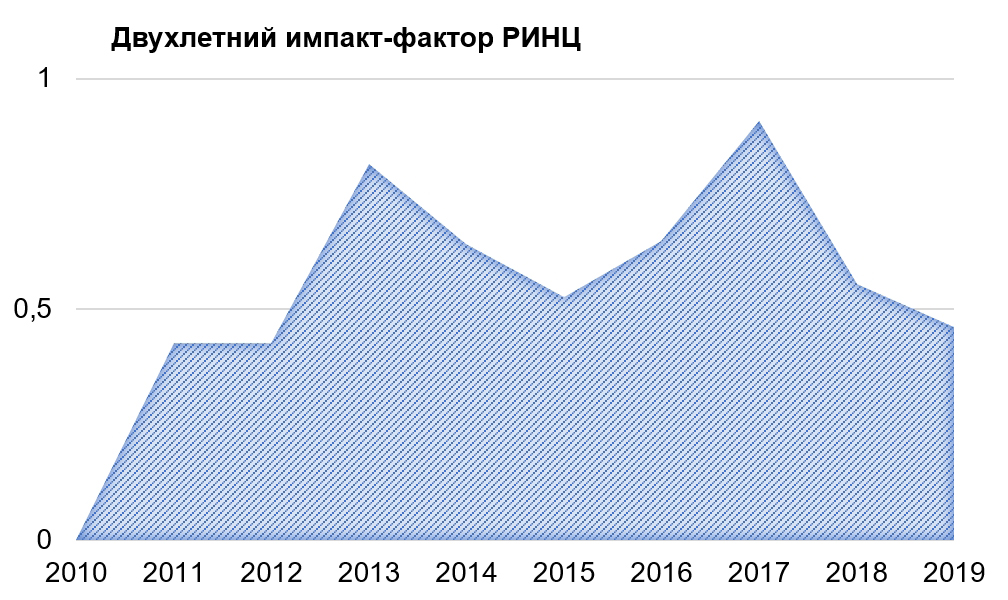
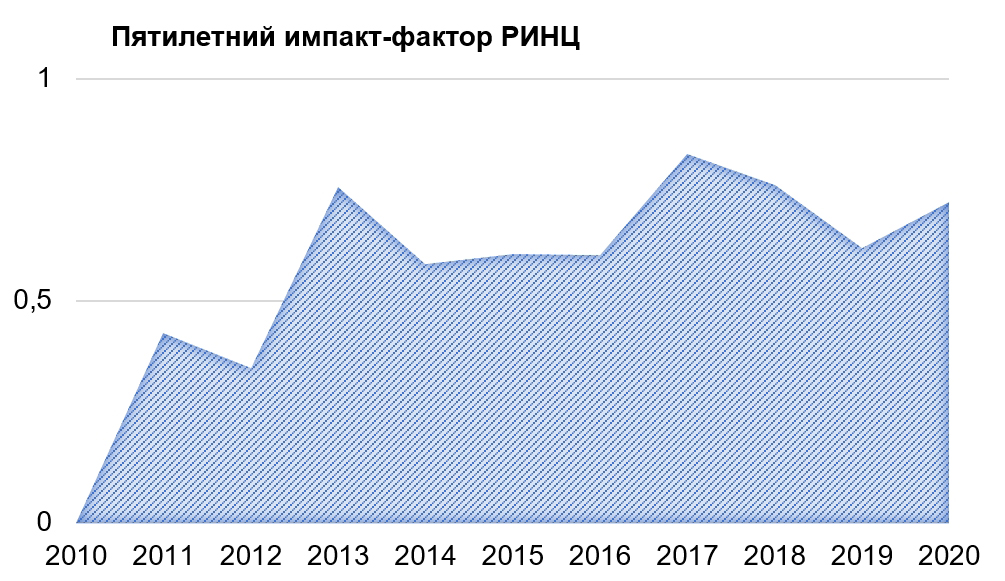
Патогенетические основы развития диабетической цистопатии
 7009
7009 Сахарный диабет (СД) – заболевание, характеризующееся нарушением обмена углеводов вследствие абсолютной или относительной недостаточности инсулина и сопровождающееся тяжелыми метаболическими нарушениями. Частоту СД среди взрослых оценивают в 6,5%, еще у 18% взрослых выявляют нарушения метаболизма глюкозы [1]. Заболеваемость СД имеет тенденцию к увеличению, причем в большей степени это относится к СД 2 типа. Значимость СД обусловлена не только его высокой распространенностью, но и вызываемыми им тяжелыми осложнениями.
Урологические осложнения являются одними из самых частых и значимых для пациента с СД. Собственно, история изучения СД неразрывно связана с обнаружением повышенного выделения мочи больными. Уже в первых упоминаниях о СД, которые обнаружили в трудах древнекитайских и древнеиндийских целителей 4-го тысячелетия до нашей эры, ведущим признаком заболевания считалось обильное мочеотделение. На полиурию в качестве ведущего симптома СД указывали великие врачи древности и средневековья – Гиппократ, Цельс, Гален, Авиценна, Парацельс. В этот период было хорошо известно о нарушениях мочеиспускания у больных СД, а их единственной причиной длительное время считали повышенное выделение мочи.
О расстройствах мочеиспускания, как самостоятельных осложнениях СД, впервые сообщил французский врач Шарль-Якоб Маршал де Кальви в 1864 году. Помимо подробного описания этих нарушений, он впервые указал на возможную роль в их развитии поражения нервной системы [2]. В 1935 году W.R. Jordan и H.H. Crabtree предположили, что диабетические дисфункции нижних мочевыводящих путей являются проявлениям автономной диабетической нейропатии и осложняют декомпенсированный или плохо компенсируемый СД [3]. Длительное время, вплоть до 60-х годов 20 века, в качестве ведущих урологических осложнений СД рассматривали инфекционно-воспалительные осложнения – пиелонефрит и цистит, а не расстройства мочеиспускания. Только с началом использования уродинамических методов обследования больных была выявлена значительно большая, чем ранее предполагали, частота диабетических нарушений мочеиспускания. В 1976 году датский уролог C. Frimodt-Mollerописал три уродинамических признака диабетических расстройств мочеиспускания – уменьшение чувствительности мочевого пузыря, увеличение емкости мочевого пузыря и нарушение сократимости детрузора с увеличением количества остаточной мочи – и предложил использовать термин «диабетическая цистопатия» [4]. Под этим термином понималось нарушение функции нижних мочевыводящих путей у больных СД, обусловленных его наличием.
На сегодняшний день накоплен большой объем информации относительно клинического течения и уродинамических проявлений диабетической цистопатии. Эти данные позволяют утверждать, что диабетическая цистопатия не сводится только к указанным выше трем «классическим» признакам. У большого числа больных на первый план выступает симптоматика гиперактивности мочевого пузыря. В настоящее время преобладает точка зрения, что ирритативные и обструктивные симптомы, являющиеся проявлением повышенной и сниженной сократительной активности детрузора при СД, являются двумя последовательными этапами развития проявлениями диабетической цистопатии. В 2009 году F. Daneshgariи соавт., основываясь на результатах экспериментальных и клинических исследований, предложили гипотезу, позволяющую объяснить разнообразие клинических проявлений диабетической цистопатии [5]. Согласно этой гипотезе на ранней стадии диабетической цистопатии появляются симптомы гиперактивности мочевого пузыря. Причины их появления – инициированная диабетической полиурией гипетрофия детрузора. Постепенно, по мере накопления токсических метаболитов и прогрессирования ангиопатии, происходит декомпенсация детрузора, клинически проявляющаяся классическими признаками диабетической цистопатии – гипоили аконтрактильностью детрузора и затруднением оттока мочи из мочевого пузыря. Декомпенсированная стадия диабетической цистопатии может сопровождаться перерастяжением мочевого пузыря, вторичной мочевой инфекцией, нарушением функции почек
Патогенез связанных с СД осложнений со стороны нижних мочевыводящих путей разнообразен и на сегодняшний день до конца не ясен. При этом полагают, что их развитие связано с микрососудистыми поражениями, сходными с таковыми при ретинопатии, нефропатии и периферической нейропатии [6]. Описаны четыре вероятных механизма развития осложнений СД [7]. Во-первых, при повышении уровня внутриклеточной глюкозы увеличивается содержание конечных продуктов гликозилирования, которые, связываясь с белками, ускоряют развитие атеросклероза, способствуют гломерулярной и эндотелиальной дисфункции. Во-вторых, при гипергликемии некоторое количество глюкозы превращается в сорбитол. Последний действует как тканевой токсин и может явиться фактором патогенеза ретинопатии, нефропатии, нейропатии, катаракты, поражения аорты. Втретьих, при повышенном содержании глюкозы в крови увеличивается образование диацилглицерола, приводящего к активации изоформ протеина киназы С, изменяющей внеклеточный матрикс протеинов вокруг эндотелиальных клеток и нейронов. В-четвертых, гипергликемия изменяет экспрессию генов TGFb, играющих важную роль в инициации пролиферативных нарушений в различных тканях. Гетерогенность патогенетических факторов является, по-видимому, одной из причин разнообразия клинических проявлений диабетических дисфункций мочевыводящих путей. На сегодняшний день не до конца ясно, все ли эти указанные выше патогенетические механизмы равнозначны, однако не подлежит сомнению, что в основе осложнений СД лежат именно микрососудистые поражения.
В мочевом пузыре микроангиопатия ухудшает кровоснабжение его стенки, вызывая ишемию и способствуя повреждению интрамуральных нервных окончаний и нарушению функции детрузора и уротелия [8]. Негативный эффект диабетической ангиопатии усиливают повышенный гиперосмолярный диурез и развивающиеся метаболические нарушения [8, 9]. Таким образом, именно диабетическая ангиопатия вызывает нарушения, непосредственно ответственные за развитие расстройств мочеиспускания – периферическую нейропатию, патологические изменения детрузора и дисфункции уротелия [6].
ДИАБЕТИЧЕСКАЯ ПЕРИФЕРИЧЕСКАЯ НЕЙРОПАТИЯ
Под диабетической нейропатией понимают симптомы периферической нервной дисфункции у пациентов с СД, у которых исключены другие возможные причины поражения нервной системы. Диабетическая нейропатия является самой частой периферической нейропатией [11]. Клинически значимую диабетическую нейропатию выявляют примерно у 10% больных СД [12]. Частота диабетической нейропатии не зависит от пола пациента, но связана с продолжительностью СД. Сообщают, что диабетическую нейропатию выявляют у 50% больных с длительностью СД 25 лет [11].
Патогенез диабетической нейропатии изучен достаточно подробно. Доказано, что ее развитие связано с диабетической микроангиопатией – поражением vasa nervorum, мелких кровеносных сосудов, питающих нервные волокна [13]. Ведущую роль в этом играют метаболические нарушения – накопление сорбитола, снижение содержания миоинозитола, уменьшение синтеза НАДФ-Н оксидазы (клеточный мембрано-связанный мультимолекулярный ферментный комплекс) и глутатиона, усиление перекисного окисления липидов, неферментное гликозилирование белков [14, 15]. При гипергликемии многие биохимические процессы приводят к избыточному образованию свободных радикалов, которые нарушают функцию эндотелия, и развитию эндоневральной гипоксии. Активность собственной антиоксидантной системы при СД снижена. Нарушение баланса антии прооксидантных систем вызывает формирование феномена «оксидативного стресса», при котором угнетается синтез оксида азота (NO) – основного регулятора тонуса сосудистой стенки, и происходит активация ядерного фактора (NF-kB), что инициирует выделение субстанций, в частности, эндотелина-1, еще более ухудшающих кровоток [16]. Доказано, что снижение синтеза NO-короткоживущего эндотелиального фактора в условиях внутриклеточного дефицита НАДФ-Н и является основной причиной нарушения микроциркуляции в питающих нервы сосудах.
Диабетическая нейропатия проявляется повреждением как афферентных, так и эфферентных нервных волокон. Для нее характерны дегенерация нейронов, наличие нейронов с вакуолями и гранулярными депозитами, демиелинизация нервных волокон n. vagus и nn. splanchnici, фрагментация и появление участков уплотнения в тазовых постганглионарных симпатических аксонах [17]. Уродинамические и нейрофизиологические исследования с определением порога чувствительности мочевого пузыря у больных СД показали, что практически у всех пациентов с начальными проявлениями диабетической цистопатии имеются нарушения функции Aδ – и С-афферентных нервных волокон стенки мочевого пузыря [9, 18, 19]. Известно, что активация интрамуральных афферентных Aδ-волокон при наполнении и растяжении мочевого пузыря является наиболее важной сенсорной функцией для инициации мочеиспускания. Уменьшение чувствительности вызывает переполнение мочевого пузыря и приводит к перерастяжению его стенки. Роль нарушений со стороны C-волокон мочевого пузыря в развитии диабетической дисфункции мочевого пузыря является другим важным направлением исследований. В нескольких проведенных исследованиях не было обнаружено прямых доказательств наличия связи между нейропатией С-волокон и развитием диабетической цистопатии [20, 21]. Возможно, это связано с тем, что обычные уродинамические исследования и «ice water» тест не обладают достаточной чувствительностью [21]. Только сравнительно недавно данная гипотеза нашла свое подтверждение при использовании внутрипузырного теста пороговой чувствительности [22]. Получены данные, свидетельствующие о наличии достоверной связи между уродинамическими показателями и повреждением Aδи C-волокон в мочевом пузыре женщин с диабетической гипоактивностью детрузора, а сама нейропатия Aδи C-волокон может быть основной причиной расстройств мочеиспускания на ранних стадиях СД [22].
Длительное время считалось, что повторяющиеся перерастяжения мочевого пузыря являются основной причиной снижения его сократительной активности на начальных стадиях диабетической цистопатии [23]. Однако проведенные в последнее десятилетие исследования показали, что патогенез снижения контрактильности детузора при СД является более сложным и важная роль в нем принадлежит фактору роста нервов NGF [6, 24]. NGF является нейротрофином, необходимым для дифференцировки нервной ткани, регенерации нервных волокон и поддержания нормальной функции нейронов [25]. Концентрация NGF в мочевом пузыре коррелирует с клиническими проявлениями дисфункций нижних мочевыводящих путей [26]. Гипергликемия способствует снижению синтеза нейротрофических факторов в аксонах и глиальных клетках, что приводит к нарушению аксонального транспорта, а также к гибели клеток нейроглии (шванновских клеток). Уменьшение синтеза NGF в мочевом пузыре или нарушение транспорта NGF к ганглиям люмбосакральных дорсальных корешков рассматривают как одну из причин диабетической нейропатии и цистопатии [27]. В эксперименте обнаружено, что снижение сократимости детрузора связано с дефицитом аксонального транспорта NGF в афферентных проводящих путях стенки мочевого пузыря. Более того, генная терапия, увеличивающая экспрессию NGF в Aδ-волокнах афферентных волокнах, усиливала сократительную активность детрузора [28]. Отмечено, что в начальном периоде развития диабетической цистопатии, проявляющимся гипертрофией мышечных элементов детрузора и повышением активности сенсорных нейронов, уровень NGF в ткани мочевого пузыря увеличивается, но в последующем, по мере декомпенсации, содержание NGF в мочевом пузыре и сенсорных нервах снижается [5]. Уменьшение экспрессии NGF при СД сопровождается снижением содержания субстанции P [29].
Весьма важное клиническое значение имеет наблюдающееся при СД повреждение эфферентных нервных волокон. Нарушение эфферентной функции мочевого пузыря у пациентов с СД, по-видимому, вызывает детрузорную гиперактивность с нарушенной сократимостью. СД может вызывать демиелинизацию и аксональную дегенерацию эфферентных нервных волокон мочевого пузыря и приводить к уменьшению ацетилхолинэстеразной активности [30]. Указанные изменения можно охарактеризовать как парциальную автономную денервацию. Наряду с этим при диабетической цистопатии отмечают активацию M2-холинорецепторов и снижение скорости холинергической передачи [31, 32]. Повышение активности M2-холинорецепторов тормозит релаксацию мочевого пузыря и способствует развитию гиперактивности детрузора.
Необходимо отметить, что другими причинами развития гиперактивности детрузора у больных СД могут быть диабетические поражения ЦНС. Детрузорная гиперактивность может развиться у больных со связанным с СД острым нарушением мозгового кровообращения. Yamaguchi и соавт. проводили МРТ головного мозга 32 пациентам с диабетической гиперактивностью детрузора и выявили у них высокую (76%) частоту множественных церебральных инфарктов. Авторы сделали вывод о том, что в развитие гиперактивности детрузора при СД могут быть вовлечены не только периферические, но и центральные механизмы, имеющие, однако, единую природу – диабетическую васкулопатию [21]. У таких пациентов также самым частым проявлением дисфункции мочевого пузыря является детрузорная гиперактивность с нарушенной сократимостью.
В исследовании С. Yi и соавт. показано, что при экспериментальном стрептозотоцин-индуцированном СД у животных к 8 неделе после инициации болезни наблюдается уменьшение чувствительности мочевого пузыря, увеличение его емкости, уменьшение сократительной активности и увеличение количества остаточной мочи [33]. Авторы связали эти нарушения со снижением содержания в мочевом пузыре и дорсальных ганглиях пептида, связанного с геном кальцитонина (ПСГК) – неадренергического, нехолинергического нейротрансмиттера. Известно, что ПСГК играет важную роль в сенсорной функции мочевого пузыря, обеспечивая ощущение его наполненности, а снижение уровня ПСГК выявляют при атонии мочевого пузыря.
ДИАБЕТИЧЕСКИЕ ИЗМЕНЕНИЯ ДЕТРУЗОРА
При СД выявляют морфологические изменения в детрузоре. При этом данные о характере этих изменений существенно различаются. Одни авторы указывают на уменьшение величины межклеточных контактов между миоцитами, повышение возбудимости мембран миоцитов, изменение плотности, распределения и состава рецепторов на поверхности гладкомышечных клеток, то есть признаков, характерных для гиперактивности детрузора [6]. Результаты других исследований, наоборот, свидетельствуют о морфологических проявлениях гипоактивности детрузора – увеличении межклеточных промежутков между миоцитами и дегенерации нервных волокон, причем степень патологических изменений увеличивалась при повышении периода со времени инициации СД [34].
Таким образом, результаты морфологических исследований противоречивы и зависят, по-видимому, от длительности СД.
При диабетической цистопатии происходит изменение плотности и распределения рецепторов на миоцитах детрузора. Y.C. Tong и соавт. сообщили, что при индукции СД в детрузоре животных в течение 2 недель происходит увеличение плотности М2-холинорецепторов на 70% [35]. Эти данные совпадают с результатами, полученными при изучении нейрогенных дисфункций мочевыводящих путей другой природы. Известно, что в нормальном мочевом пузыре соотношение между М2и М3-субтипами холинорецепторов составляет примерно 4:1, однако в сокращении детрузора задействованы, главным образом, М3-холинорецепторы. У больных с нейрогенными дисфункциями мочевыводящих путей существенно возрастает роль М2-холинорецепторов. В денервированных мочевых пузырях плотность М2-холинорецепторов увеличивается на 60% по сравнению с контролем, в то время как плотность М3-холинорецепторов не изменяется, и, кроме того, значимо увеличивается аффинитет М2и М3-холинорецепторов к ацетилхолину [36]. K.J. Pak и соавт. показали, что в детрузоре крыс с экспериментальным СД уменьшается М3-индуцированная и повышается М2-индуцированная сократимость миоцитов [37]. Сходные изменения были выявлены ранее при гипертрофии детрузора вследствие инфравезикальной обструкции [38] и у больных после спинальной травмы [39]. В этой связи применение антихолинергических препаратов у таких больных в обычной дозе малоэффективно, зачастую требуется назначение двойных, а то и тройных доз лекарственных средств или комбинированной терапии несколькими антихолинергическими препаратами.
Повышение холинергического ответа в диабетическом детрузоре может происходить вследствие повышения чувствительности М-холинорецепторов к ацетилхолину, повышения высвобождения ацетилхолина или комбинации этих двух факторов. Высказана гипотеза, что повышение высвобождения ацетилхолина может быть самым первым признаком дегенеративных изменений в холинергических нервах, вызванных СД, и что эти патологические изменения могут вызывать утрату нервами способности контролировать количество выделяемых нейротрансмиттеров [40]. Альтернативным объяснением повышения холинергического ответа может быть гипотетический компенсаторный механизм холинергического компонента в ответ на недостаток нехолинергического компонента моторной передачи в диабетическом детрузоре. Можно допустить, что холинергический и не-холинергический компоненты действуют сообща, и дефицит одного из них (в данном случае, нехолинергического) ведет к компенсаторному повышению другого – холинергического [40].
Для диабетической цистопатии характерны изменения нейромышечной трансмиссии. У здорового человека сокращение детрузора практически полностью обеспечивается холинергическим и пуринергическим компонентами, причем вклад последнего не превышает 5% [41]. При диабетической цистопатии уменьшается доля холинергической нервно-мышечной передачи и, наоборот, существенно увеличивается вклад АТФ-опосредованной пуринергической нейротрансмиссии [42]. Сходные с СД изменения на уровне нервномышечной передачи в детрузоре происходят на ранних стадиях инфравезикальной обструкции. Наряду с изменением плотности холинорецепторов в миоцитах детрузора наблюдают изменение экспрессии адренорецепторов. Y. Kubota и соавт. выявили усиление ß1-адренорецептор-опосредованного расслабления детрузора крысы через 8-10 недель после индукции СД первого типа стрептозотоцином [43].
C. Wang и соавт. в эксперименте на крысах с стрептозотоцин-индуцированным СД изучали механические свойства стенки мочевого пузыря и выявили повышение массы мочевого пузыря и его емкости по сравнению с контролем. Авторы обнаружили увеличение растяжимости стенки мочевого пузыря и предположили, что полиурия ответственна главным образом за «ранние» изменения механических свойств стенки мочевого пузыря, а связанные с СД метаболические нарушения вызывают следующие за ними «поздние» изменения [44].
Таким образом, анализ имеющейся информации свидетельствует о сложных и иногда разнонаправленных процессах в диабетическом детрузоре. Обобщив эти данные, можно предположить, что изменения детрузора при СД проходят две фазы развития. Во время первой фазы сократительная способность миоцитов увеличивается, что уродинамически проявляется развитием гиперактивности детрузора. Вторая фаза характеризуется декомпенсацией мышечного слоя мочевого пузыря, снижением его сократительной активности вплоть до аконтрактильности, что клинически выражается в затруднении мочеиспускания, увеличении количества остаточной мочи, а иногда и задержкой мочи.
ДИСФУНКЦИИ УРОТЕЛИЯ
В последнее время к изучению роли уротелия в развитии различных дисфункций мочевыводящих путей уделяется повышенное внимание. Доказано, что уротелий является не объектом, а субъектом регуляции мочеиспускания. Уротелий способен воспринимать различные стимулы и выделять биологически активные вещества, воздействуя на мышечные и нервные элементы в нижележащих слоях стенки мочевого пузыря. Основными функциями уротелия считают барьерную и сенсорную. Для обеспечения последней уротелий обладает способностью воспринимать и высвобождать множество медиаторов, воздействующих на афферентные нервные окончания, и таким образом участвовать в развитии различных диабетических дисфункций мочевого пузыря. Барьерную функцию уротелия поддерживают тесные межклеточные соединения и наличие специальных липидных и протеиновых (уроплакин) молекул в его поверхностном слое. Местные повреждения (инфекция, механическая и химическая травма) могут нарушать барьерную функцию уротелия. В результате различные субстанции из мочи могут проникать в подлежащие ткани, усиливать афферентную стимуляцию и клинически проявляться учащением мочеиспускания, императивными позывами и болями во время наполнения и опорожнения мочевого пузыря [45].
На сегодняшний день известно, что уротелий содержит пуринергические рецепторы P2Y и P2X и способен выделять медиаторы NO и ATФ [46]. Активация пуринергических рецепторов во время растяжения мочевого пузыря играет сигнальную роль в мочевом пузыре. Установлено, что повышение внутрипузырного давления и растяжение мочевого пузыря стимулирует выделение уротелием АТФ, которые связываются с пуринергическими P2Xи, возможно, с P2Y-рецепторами клеток поверхностного слоя уротелия [47]. Предполагают, что диабетическая полиурия, сопровождающаяся растяжением мочевого пузыря, на ранних стадиях диабетической цистопатии приводит к увеличению чувствительности мочевого пузыря за счет именно этих уротелиальных механизмов. Кроме того, известно, что частота бактериурии и инфекционно-воспалительных заболеваний мочевыделительной системы у больных СД повышается. Инфицирование и травматизация стенки мочевого пузыря способствует повреждению уротелия, что сопровождается выделением большого количества АТФ и может вызывать ирритативные симптомы и боль. Весьма интересные наблюдения приведены в работе А. Munoz и соавт., показавших, что гиперактивность детрузора при диабетической цистопатии сопровождается повышением синтеза ATФ уротелием, а гипоактивность детрузора – увеличением высвобождения NO при нормальном уровне АТФ [48]. Уротелий также синтезирует простагландины F2α и Е2, являющиеся важными медиаторами функции мочевого пузыря в норме и при патологических состояниях. В эксперименте установлено, что при СД синтез простагландинов уротелием нарушается, что, возможно, также играет роль в патогенезе диабетической цистопатии [49].
Суммируя известные на сегодняшний день данные, можно утверждать, что роль уротелия в развитии и прогрессировании дисфункций нижних мочевыводящих путей у больных СД весьма велика, однако изучена еще недостаточно и требует дальнейших исследований.
Таким образом, согласно современным представлениям, ведущими патогенетическими факторами диабетической цистопатии являются нейропатия, поражение детрузора и дисфункция уротелия. При этом появление этих нарушений обусловлено диабетической ангиопатией, являющейся, по существу, основой развитии диабетической цистопатии.
ЛИТЕРАТУРА
1. Cowie CC, Rust RF, Byrd-Holt DD, Eberhardt MS, Flegal KM, Engelgau MM Prevalence of diabetes and impaired fasting glucose in adults in the U.S. population: National health and Nutrition Examination Survey 1999-2002 // Diabetes Care. 2006. Vol. 29. Р. 1263.
2. Marchal de Calvi Ch-J Recherches sur les accidents diabétiques et essai d'une théorie générale de diabète. Asselin und Labé. 1864. 658 p.
3. Jordan WR, Crabtree HH. Paralysis of the Bladder in Diabetic patients. // Arch Inter Medicine. 1935. Vol.55. P. 17-25.
4. Frimodt-Moller C. Diabetic cystopathy. I: A clinical study of the frequency of bladder dysfunction in diabetics. // Dan Med Bull. 1976. Vol. 23. P. 267-278.
5. Daneshgari F, Liu G, Birder L, Hanna-Mitchell AT, Chacko S. Diabetic bladder dysfunction: current translational knowledge // J Urol. 2009. Vol. 182. Р. 18-26.
6. Yoshimura N, Chancellor MB., Andersson KE. Recent advances in understanding the biology of diabetes-associated bladder complications and novel therapy. // BJU Int. 2005. Vol. 95. P. 733–738.
7. Jensen D, Klevmark B. Systemic illnesses (diabetes mellitus, sarcoidosis, alcoholism, and porphyrias.// In: Textbook of the Neurogenic Bladder, 2nd Edition. [ed. J. Corcos, E. Schick]. Informa UK Ltd, UK. 2008. – P. 229-239. 8. Yamaguchi O, Nomiya M, Andersson KE. Functional consequences of chronic bladder ischemia. // Neurourol Urodyn. 2014. Vol.33, N 1. P. 54-58.
9. Lee WC, Wu HP, Tai TY, Liu SP, Chen J, Yu HJ. Effects of diabetes on female voiding behavior. // J Urol. 2004. Vol.172, N 3. P.989-992.
10. Brown JS. Diabetic cystopathy – what does it mean? // J Urol. 2009. Vol. 181, N 1. P. 13-14.
11. Bansal V, Kalita VJ, Misra UK. Diabetic neuropathy. // Postgrad Med J. 2006. Vol. 82. P. 95–100.
12. Schaumburg HH, Berger AR, Thomas PK. Diabetic neuropathy. // In: Disorders of Peripheral nerves. 2nd edn. – Philadelphia: Davis Company. 1992. P. 91-109.
13. Malik RA, Newrick PG, Sharma AK. Microangiopathy in human diabetic neuropathy: relationship between capillary abnormalities and the severity of neuropathy. // Diabetologia. 1989. Vol.32. P.92-102.
14. Аметов А.С., Строков И.А. Диабетическая полинейропатия: настоящее и будущее // Российские медицинские вести. 2001. Т.4, №1. С. 35–40. 15. Llewelyn JG, Tomlinson DR, Thomas PK. Diabetic neuropathies. // In: Peripheral Neuropathy, 4th edn. Philadelphia: Elsevier Saunders. 2005. Р.1951-1991.
16. Changolkar A, Hypolite J, Wein AJ. Decreased contractility of the detrusor smooth muscle in diabetes is associated with alteration of the intracellular reduction-oxidation (redox) state and activation of nuclear transcription factor kappa B (NF-KB). // J Urol. 2002. Vol. 167, N 4. P. 248.
17. Low PA, Dyck PJ. Pathologic studies and the nerve biopsy in autonomic neuropathies. // In: Clinical Autonomic Disorders. [ed. Low P.A.]. Boston.: Little, Brown. 1993. Р.331-344.
18. Ørstavik K, Namer B, Schmid R. Abnormal function of C-fibers in patients with diabetic neuropathy // J Neurosci. 2006. Vol. 26. P. 11287-11294.
19. Kebapci N, Yenilmez A, Efe B, Entok E, Demirustu C. Bladder dysfunction in type 2 diabetic patients // Neurourol Urodyn. 2007. Vol. 26, N 6. Р.814–819.
20. Ishigooka M, Hashimoto T, Hayami S, Suzuki Y, Ichiyanagi O, Nakad T. Thermoreceptor mediated bladder sensation in patients with diabetic cystopathy // Int Urol Nephrol. 1997. Vol. 29. Р. 551–555.
21. Yamaguchi C, Sakakibara R, Uchiyama T, Yamamoto T, Ito T, Liu Z, Awa Y, Yamamoto K, Nomura F, Yamanishi T, Hattori T. Overactive bladder in diabetes: a peripheral or central mechanism? // Neurourol Urodyn. 2007. Vol. 26, N 6. P. 807-813.
22. Lee WC, Wu HP, Tai TY, Yu HJ, Chiang PH. Investigation of urodynamic characteristics and bladder sensory function in the early stages of diabetic bladder dysfunction in women with type 2 diabetes // J Urol. 2009. Vol.181, N 1. P.198–203.
23. Hampel C, Gillitzer R, Pahernik S, Melchior S, Thüroff JW. Diabetes mellitus and bladder function. What should be considered? // Urology. 2003. Vol. 42, N 12. P. 1556-1563.
24. Lee WC, Wu CC, Wu HP, Tai TY. Lower urinary tract symptoms and uroflowmetry in women with type 2 diabetes mellitus with and without bladder dysfunction. // Urology. 2007. Vol.69. P. 685-90.
25. Levi-Montalchini R. The nerve growth factor 35 years later. // Science. 1987. Vol.237. P. 1154-1162.
26. Cruz CD. Neurotrophins in bladder function: what do we know and where do we go from here? // Neurourol Urodyn. 2014. Vol.33, N 1. Р. 39–45.
27. Brown JS, Wessells H, Chancellor MB, Howards SS, Stamm WE, Stapleton AE. Urologic complications of diabetes. // Diabetes Care. 2005. Vol. 28, N 1. P. 177–185.
28. Sasaki K, Chancellor MB, Phelan MW. Diabetic cystopathy correlates with a long-term decrease in nerve growth factor levels in the bladder and lumbosacral dorsal root Ganglian. // J Urol. 2002. Vol.168, N 3. P.1259–1264.
29. Li Y, Shi B, Wang D, Wang P, Laudon V, Zhang J, Liu Y. Nerve growth factor and substance P: expression in a rat model of diabetic bladder. // Int Urol Nephrol. 2011. Vol.43, N 1. P.109-116.
30. Van Poppel H, Stessens R, Van Damme B, Carton H, Baert L. Diabetic cystopathy: neuropathophysiologal examination of urinary bladder biopsies. // Eur Urol. 1998. Vol.15, N 1-2. P.128-131.
31. Yono M, Latifpour J, Yoshida M, Ueda S. Age-related alterations in the biochemical and functional properties of the bladder in type 2 diabetic GK rats. // J Recept. Signal Transduct Res. 2005. Vol. 25. P. 147-157.
32. Tong YC, Cheng JT, Hsu CT. Alterations of M(2)-muscarinic receptor protein and mRNA expression in the urothelium and muscle layer of the streptozotocin-induced diabetic rat urinary bladder. // Neurosci Lett. 2006. Vol. 406. P. 216-221.
33. Yi C, Wei Z, Wang H, Song T, Ding L. The effects of peptidergic nerve (calcitonin gene-related peptide, cgrp) and calcium-related contraction on the voiding dysfunction and histopathological alteration in the diabetic cystopathy rats. // 40th Annual Meeting of International Incontinence Society, Toronto, Canada. 2010. Р. 516.
34. Rizk DE, Padmanabhan RK, Tariq S, Shafiullah M, Ahmed I. Ultra-structural morphological abnormalities of the urinary bladder in streptozotocin-induced diabetic female rats. // Int Urogynecol J Pelvic Floor Dysfunct. 2006. Vol.17. P. 143-154.
35. Tong YC, Chin WT, Cheng JT. Alterations in urinary bladder M2-muscarinic receptor protein and mRNA in 2-week streptozotocininduced diabetic rats // Neurosci Lett. 1999. Vol.277, N 3. P.173-176.
36. Braverman AS, Luthin GR, Ruggieri MR. M2 muscarinic receptor contributes to contraction of the denervated rat urinary bladder. // Am J Physiol. 1998. Vol. 275, N 5. P. 1654−1660.
37. Pak KJ, Ostrom RS, Matsui M, Ehlert FJ. Impaired M3 and enhanced M2 muscarinic receptor contractile function in a streptozotocin model of mouse diabetic urinary bladder. // Naunyn-Schmied Arch Pharmacol. 2010. Vol.381. P.441–454.
38. Braverman AS, Ruggieri MR. Hypertrophy changes the muscarinic receptor subtype mediating bladder contraction from M3 toward M2. // Am J Physiol Regul Integr Comp Physiol. 2003. Vol. 285. P. 701-708.
39. Pontari MA, Braverman AS, Ruggieri MR. The M2 muscarinic receptor mediates in vitro bladder contractions from patients with neurogenic bladder dysfunction. // Am J Physiol Regul Integr Comp Physiol. 2004. Vol. 286. Р. 874–880.
40. Luheshi GN, Zar MA. The effect of streptozotocin-induced diabetes on cholinergic motor transmission in the rat urinary bladder. // Br J Pharmacol. 1991. Vol. 103. P. 1657-1662.
41. Burnstock G. Purinergic signalling in lower urinary tract: Handbook of Experimental Pharmacology I: Purinergic and Pyrimidinergic Signalling I //In: Molecular, Nervous and Urinogenitary System Function: Springer-Verlag. Berlin. 2001. Vol. 151. Р. 423-515.
42. Mumtaz FH, Lau DHW, Siddiqui EI, Morgan RJ, Thompson CS, Mikhailidis DP. Changes in Cholinergic and Purinergic Neurotransmission in the Diabetic Rabbit Bladder. // In Vivo. 2006. Vol.20, N 1. P. 1-4.
43. Kubota Y, Nakahara T, Mitani A, Maruko T, Sakamoto K, Ishii K. Augmentation of rat urina
ry bladder relaxation mediated by beta1-adrenoceptors in experimental diabetes. // Eur J Pharmacol. 2003. Vol. 467. Р. 191–195.
44. Wang CC, Nagatomi J, Toosi KK, Yoshimura N, Hsieh JH, Chancellor MB, Sacks MS. Diabetes-induced alterations in biomechanical properties of urinary bladder wall in rats. // Urology. 2009. Vol. 73, N 4. P. 911-915.
45. Birder LA, De Groat WC. Mechanisms of disease: involvement of the urothelium in bladder dysfunction. // Nat Clin Pract Urol. 2007. Vol. 4. P. 46-54.
46. Apodaca G. The urothelium: not just a passive barrier. // Traffic. 2004. Vol. 5. P. 117–128.
47. Wang CC, Lee JM, WG Ruiz WG, Balestreire EM, Bodungen M, Barrick S. ATP and purinergic receptor-dependent membrane traffic in bladder umbrella cells. // J Clin Invest. 2005. Vol. 115. P. 2412-2422.
48. Munoz A, Smith C, Boone T, Somogyi G. Overactive and underactive bladder dysfunction is reflected by alterations in urothelial ATP and NO release. // Neurochem Int. 2011. Vol. 58, N 3. P. 295–300.
49. Pinna C, Zanardo R, Puglisi L. Prostaglandin-release impairment in the bladder epithelium of streptozotocin-induced diabetic rats. // Eur J Pharmacol. 2000. Vol. 388. Р. 267-273
| Прикрепленный файл | Размер |
|---|---|
| Скачать статью | 984.32 кб |
Другие статьи выпуска




![]()
![]()