







ВВЕДЕНИЕ
Согласно современным представлениям о патогенезе мочекаменной болезни (МКБ), уролитиаз является полиэтиологическим и полипатогномоничным заболеванием [1]. Как известно, причинами литогенеза могут быть некоторые генетические мутации, сопутствующие заболевания (ожирение, гиперпаратиреоз и т.д.), инфекция мочевых путей, особенности диеты и др. [2].
В то же время, в мировой литературе достаточно часто встречается термин «идиопатический уролитиаз», чаще всего применяемый при кальций-оксалатной или кальций-фосфатной формах МКБ [3-6]. Одной из вероятных причин формирования «идиопатических» кальциевых мочевых камней может быть сопутствующая тубулопатия – наследственная и приобретенная почечная канальцевая дисфункция [7]. Изучением данного вопроса занимаются, в основном, детские урологи и нефрологи, поскольку в большинстве случаев именно в детском возрасте клинически проявляются известные в настоящее время тубулопатии.
МАТЕРИАЛЫ И МЕТОДЫ
При составлении обзора литературы использовались данные, опубликованные в базах PubMed, научной электронной библиотеке eLibrary.ru, на сайте Европейской ассоциации урологов. Поиск в базах данных проводился по ключевым словам: «уролитиаз», «urolithiasis», «почечный канальцевый ацидоз», «renal tubular acidosis», «тубулопатия», «tubulopathy». Было найдено 89 источников не старше 10 лет (опубликованные после 2012 года), которые имели отношение к теме обзора. Из них были исключены тезисы конференций, короткие сообщения, дублирующиеся публикации. После чего, исходя из актуальности данных, достоверности источников, импакт-факторов журналов и последовательности изложения материала в рукописи, непосредственно для цитирования в обзоре было отобраны 65 статей. Также при написании обзора использовались оригинальные статьи, опубликованные до 2012 года.
РЕЗУЛЬТАТЫ
Почечный канальцевый ацидоз (ренальный тубулярный ацидоз) (ПКА) является наиболее изученной тубулопатией, приводящей к МКБ. В настоящий момент выделяют 4 типа ПКА: дистальный (тип I), проксимальный (тип II), комбинированный (III тип) и гиперкалиемический (IV тип) [8]. Основной интерес для практикующего уролога представляет ПКА дистального типа, поскольку именно при данном типе формируются мочевые камни.
Одной из основных функций почек является поддержание кислотно-щелочного равновесия организма [9]. Данный процесс протекает при помощи буферных систем – физиологических жидкостей организма, сохраняющих постоянное значение рН. Действие всех буферных систем взаимосвязано. При этом одним из основных механизмов поддержания гомеостаза является гидрокарбонатная буферная система, которая составляет 53% общей буферной емкости крови. Гидрокарбонатная буферная система состоит из молекулы угольной кислоты H2CO3, являющейся источником водорода и отрицательно заряженного иона гидрокарбоната HCO3−, выполняющего роль акцептора водорода:
Н2О + СО2 « H2CO3 « Н+ + НСО3−.
Выделение СО2 происходит через легкие, ионы Н+ и HCO3− выделяются через почки [10]. Изменение концентрации Н+ на 1 ммоль приводит к изменению рН организма на 0,01. Биологический смысл буферной системы состоит в том, чтобы поддерживать постоянство рН в крови и тканях.
Почки ежедневно фильтруют около 180 л первичной мочи, содержащей в общей сложности около 4500 мэкв гидрокарбоната [11]. При необходимости почки экскретируют Н+ из кислой среды и HCO3− из щелочной [12, 13]. При снижении уровня гидрокарбоната в плазме проксимальные канальцы реабсорбируют его до концентрации 25 ммоль/л, в результате чего гидрокарбонатный буфер восстанавливается.
Любые нарушения в работе буферной системы влияют на состояние клеточных мембран, внутриклеточную передачу сигналов и метаболизм, что приводит к серьезным дисфункциям в работе организма.
ПКА характеризуется нарушением экскреции нелетучих кислот почками при сохраненной скорости клубочковой фильтрации (СКФ) [14]. При ПКА в сыворотке крови прослеживается или нормальный анионный провал (то есть разница между содержанием катионов и анионов в плазме крови составляет 12 мэкв/л) или гиперхлоремический метаболический ацидоз, вызванный неспособностью почечных канальцев удерживать бикарбонат НСО3− или секретировать ионы водорода Н+ при нормальной или умеренно нарушенной функции почек [15]. При этом необходимо подчеркнуть, что при I типе ПКА происходит именно нарушение экскреции водородных ионов в дистальном отделе нефрона (рис. 1), а проксимальный ПКА (II типа) характеризуется нарушением реабсорбции бикарбонатов (НСО3−) в проксимальных канальцах [16].
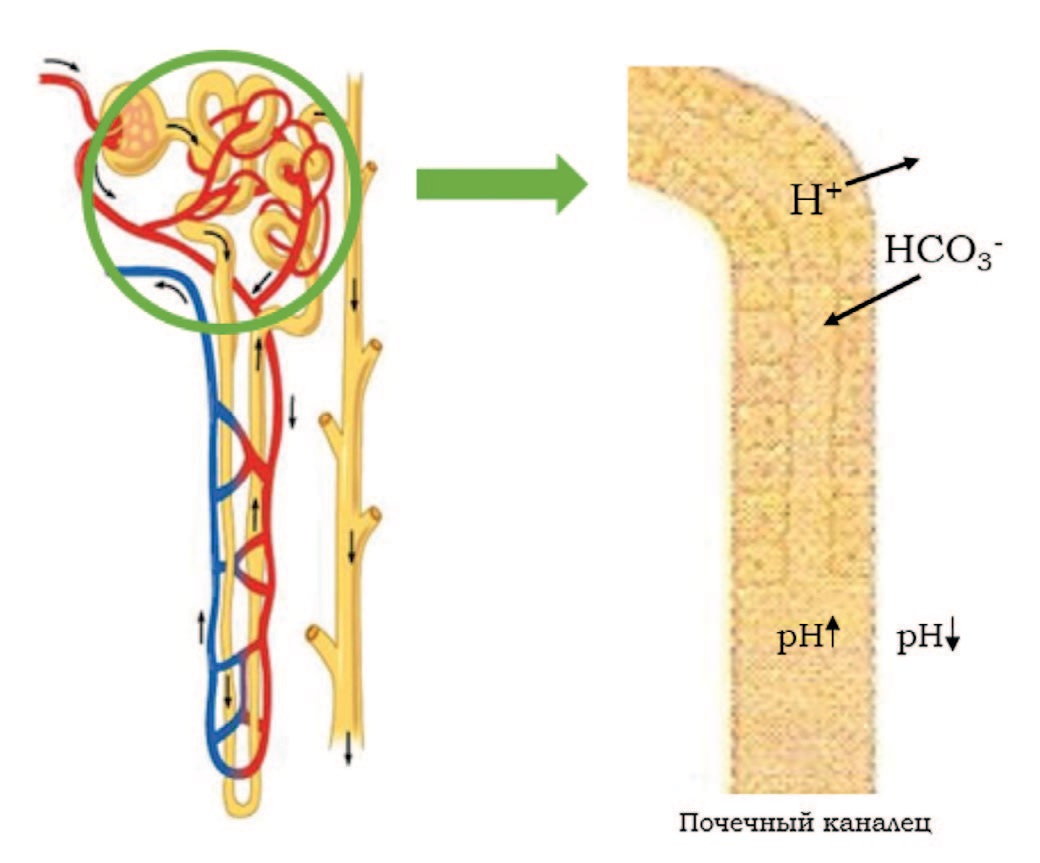
Рис. 1. Нарушение метаболизма ионов водорода Н+ и бикарбоната НСО3- в почечных канальцах при ПКА дистального типа [23]
Fig. 1. Impaired metabolism of hydrogen ions H+ and bicarbonate HCO3- in the renal tubules in distal-type RTA [23]
Согласно данным мировой литературы, частота уролитиаза у пациентов с ПКА 1 типа варьирует от 7% до 52-77% [17, 18]. В то же время у пациентов с МКБ ПКА 1 типа встречается в 0,3 – 8% случаев [19].
Необходимо отметить, что при дистальном типе ПКА выделяют так называемую неполную форму ПКА [20]. Особенности неполного ПКА дистального типа заключаются в том, что у пациентов отсутствует метаболический ацидоз и/или ацидемия. Основным лабораторным проявлением неполной формы ПКА является неспособность почек снижать pH мочи ниже 5,5 [21]. При неполной форме ПКА дистального типа у пациентов также протекают процессы камнеобразования в почках, как и у больных с полной формой ПКА. Необходимо подчеркнуть, что до сих пор стоит вопрос, является ли неполная форма ПКА самостоятельным заболеванием или вариантом проявления ПКА.
В Швейцарии проведено исследование, в котором изучена вероятность наличия неполной формы ПКА дистального типа у мужчин с рецидивирующим течением кальциевого уролитиаза. Согласно полученным данным у 7% пациентов с кальциевыми камнями (10 больных из 150 исследуемых) была диагностирована неполная форма ПКА [22].
Известно, что ПКА может быть как первичным (наследственным), так и вторичным (приобретенным) [23].
Как правило, диагностированный в детстве ПКА связан с генетическими мутациями. Наследственный тип ПКА проявляется клинически, в основном, в раннем детстве, хотя у лиц с аутосомно-доминантной мутацией гена SLC4A1 может возникать и в более позднем возрасте. Наследственные формы ПКА обусловлены мутациями, в основном, в следующих генах: SLC4A1, ATP6V1B1, ATP6V0A4 (табл. 1) [24].
Таблица 1. Основные типы мутаций при дистальной форме ПКА
Table 1. The main types of mutations in the distal form of RTA
| Ген Gene |
Тип наследования Inheritance type |
Время манифестации ПКА RTA manifestation time |
Клинические проявления Clinical manifestations |
|---|---|---|---|
| SLC4A1 | аутосомно-доминантный autosomal dominant |
Детство, подростковый возраст Childhood, adolescence |
МКБ, нефрокальциноз, задержка роста, анемия, боль в левой половине живота, холецистит, деформация скелета KSD, nephrocalcinosis, growth retardation, anemia, left abdominal pain, cholecystitis, skeletal deformity |
| ATP6V1B1 | аутосомно-рецессивный autosomal recessive |
Раннее детство Early childhood |
МКБ, нефрокальциноз, тугоухость, рвота, полиурия KSD, nephrocalcinosis, hearing loss, vomiting, polyuria |
| ATP6V0A4 | аутосомно-рецессивный autosomal recessive |
Подростковый возраст, позднее детство Adolescence, late childhood |
МКБ, нефрокальциноз, мышечная слабость, рахит, остеомаляция и деформация скелета KSD, nephrocalcinosis, muscle weakness, rickets, osteomalacia and skeletal deformity |
| FOXI1 | аутосомно-рецессивный autosomal recessive |
Нет данных No data |
МКБ, нефрокальциноз, тугоухость, мужское бесплодие KSD, nephrocalcinosis, hearing loss, male infertility |
| WDR72 | аутосомно-рецессивный autosomal recessive |
Нет данных No data |
МКБ, нефрокальциноз, нарушения амелогенеза KSD, nephrocalcinosis, disorders of amelogenesis |
Ген SLC4A1 кодирует информацию о структуре 2 изомеров белка: анион-транспортного белка Cl–/HCO3- 1 в эритроцитах (полоса 3/eAE1) и укороченную форму анионообменного транспортера 1, который экспрессируется в α-интеркалированных клетках почек (kAE1). Помимо ПКА дистального типа мутация в гене SLC4A1 может вызывать наследственный микросфероцитоз (болезнь Минковского-Шоффара) [25]. При мутации гена SLC4A1 ПКА возникает вследствие нарушений ацидификации мочи.
Достаточно важным является тот факт, что у пациентов с мутацией гена SLC4A1 помимо клинических проявлений ПКА дистального типа может также встречаться картина наследственного микросфероцитоза (бледность или желтушность кожных покровов, слизистых оболочек вследствие анемии, гемолиза, боли в левой половине живота из-за увеличения селезенки, холецистит, деформация скелета, и др.) [26, 27]. Сообщается, что сочетание клинической картины двух заболеваний, в основном, встречается при мутации гена SLC4A1 [28]. При этом необходимо помнить, что наследственный микросфероцитоз может быть связан не только с мутацией гена SLC4A1, но и с мутациями в других генах [25].
Доказано, что мутация гена SLC4A1 наследуется по аутосомно-доминантному типу [29, 30]. Так обнаружено, что у пациентов с дистальной формой ПКА и наследственным микросфероцитозом последовательность нуклеотидов TGC в гене SLC4A1 заменена на TGG в экзоне 13, а последовательность GGC заменена на GAC в экзоне 17. Подобные мутации были обнаружены и у родителей пациентов [31]. При этом сообщается, что наследование мутации происходит в основном по аутосомно-доминантному типу, хотя встречается и аутосомно-рецессивное наследование [28].
Интересным является тот факт, что мутации гена SLC4A1 гораздо чаще встречаются среди населения тропических стран, в частности, Тайланда, Малайзии, Филиппин и Папуа-Новой Гвинеи. Предполагается, что изменения эритроцитов, вызванные этой мутацией, могут защищать от малярии, которая достаточно часто встречается в тропиках [32].
Другой ген, мутации которого могут приводить к ПКА дистального типа, ATP6V1B1 кодирует информацию о структуре части белкового комплекса (субъединица бета-1), известного как вакуолярная Н(+)-АТФаза (V-АТФаза) (рис. 2). Данный белок является протонным насосом внутриклеточных мембран. Протонная VАТФаза была обнаружена в мембранах, которые ограничивают различные внутриклеточные компартменты с более кислым по сравнению с цитоплазмой содержимым [33]. Таким образом, V-АТФаза создает кислую среду внутри литических вакуолей и секреторных везикул. Кислая среда необходима для создания оптимальных условий работы гидролитических ферментов. В связи с этим, она является ключевым регулятором огромного числа биологических процессов, таких как деградация белков, автофагия, обмен мембран (мембранный трафик, везикулярная секреция), вторичный транспорт через внутриклеточные мембраны [34, 35].
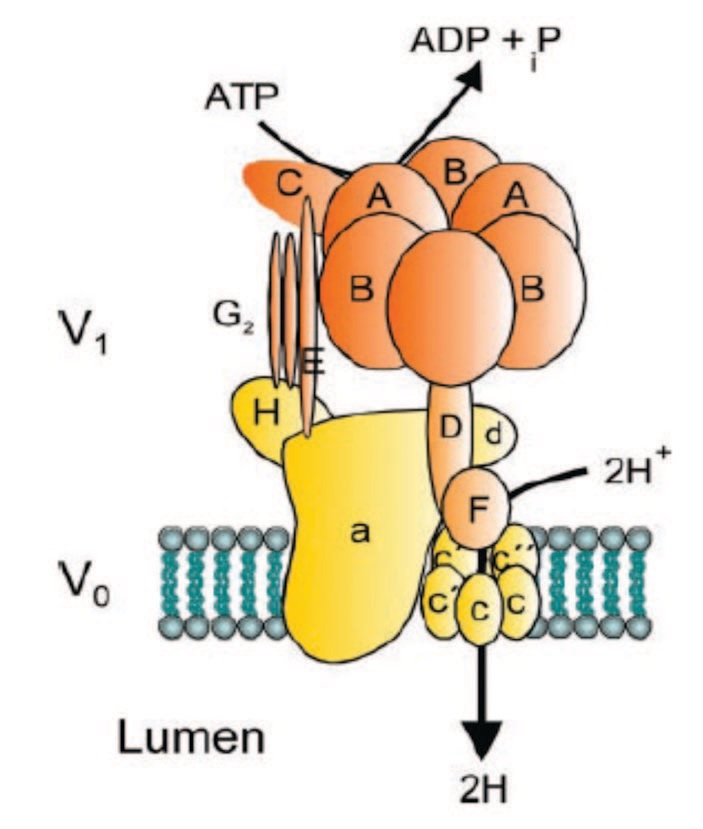
Рис. 2. Структурная модель вакуолярной Н-АТФазы. Цитозольный домен V1 состоит из субъединиц A-H, а мембраносвязанный домен V0 состоит из субъединиц a-d с несколькими изоформами субъединицы c [37]
Fig. 2. Structural model of vacuolar H-ATPase. The cytosolic V1 domain consists of A-H subunits, and the membrane-bound V0 domain consists of a-d subunits with several isoforms of the c subunit [37]
Вакуолярная Н(+)-АТФаза экспрессируется в плазматической мембране клеток почки практически по всей длине нефрона. В дистальных отделах нефрона VАТФаза отвечает за экскрецию ионов Н+ нефрона, в проксимальных канальцах V-АТФаза способствуют реабсорбции бикарбоната [36].
Опубликованы работы, в которых сообщается, что мутации гена ATP6V1B1 могут проявляться фенотипически также в виде тугоухости и нарушений слуха [38]. Исходя из собственного клинического опыта, можем сказать, что в клинической практике встречаются пациенты с врожденной наследственной тугоухостью и рецидивирующим течением уролитиаза. Как правило, семейный анамнез у данной категории больных отягощен, а манифестация МКБ зачастую происходит в детстве или в подростковом возрасте, при этом тугоухость носит врожденный характер.
Доказано, что мутация гена ATP6V1B1 наследуется по аутосомно-рецессивному типу (табл. 1) [29, 39, 40].
Ген ATP6V0A4, так же, как и ген ATP6V1B1 кодирует информацию о структуре фермента V-АТФазы (субъединица альфа-4). Соответственно, фенотипические нарушения, характерные для мутации гена ATP6V1B1 свойственны и для пациентов с мутацией гена ATP6V0A4. Впервые F.E. Karet и соавт. описали мутацию вышеназванного гена в 1999 году [41]. Дальнейшие работы подтвердили верность их заключений [29, 39, 40].
В таблице 1 представлены основные фенотипические проявления мутаций, приводящих к дистальной форме ПКА.
Сообщается также, что мутации генов FOXI1 и WDR72 могут приводить к дистальной форме ПКА.
В 2018 году S. Enerbäck и соавт. опубликовали данные о мутации гена FOXI1, проявляющейся у пациентов с гиперхлоремией, дистальной формой ПКА и нейросенсорной тугоухостью, при этом больные являлись представителями двух семей [42]. Указанная мутация FOXI1 является точечной мутацией (миссенсмутацией). Ген FOXI1 кодирует информацию о структуре ионообменников AE1 и AE4, локализующихся в почках, а также отвечает за строение субъединиц А, В1, Е2 и а4 фермента вакуолярной Н-АТФазы [43, 44].
Исследования на мышах показали, что делеция гена FOXI1 может приводить к глухоте и бесплодию у самцов [44]. Сообщается, что у мышей ген FOXI1 экспрессируется в почках, эпителии эндолимфатического протока перепончатого лабиринта и в эпителии яичка [43, 45].
Учитывая вышеуказанные данные, в настоящий момент высказывается предположение, что у пациентов мужского пола мутация гена FOXI1 фенотипически может проявляться не только в виде ПКА дистального типа и тугоухости, но и в виде бесплодия.
Опубликованы работы, утверждающие, что мутация гена WDR72 приводит к ПКА дистального типа и нарушениям в процессе амелогенеза – образования структуры эмали зубов. Сообщается, что проведенный скрининг пациентов показал, что у некоторых больных с мутацией гена WDR72 был диагностирован метаболический ацидоз и выявлены нарушения подкисления мочи – основные признаки ПКА дистального типа. Необходимо подчеркнуть, что мутация гена WDR72 наследуется по аутосомно-рецессивному типу. Данные о наличии признаков нарушения слуха у данной категории больных отсутствуют [46, 47].
В настоящее время изучается возможность влияния мутации генов SLC12A7 (белок K+/Cl- – котранспортер KCC4), SLC26A7 (Cl- /HCO3- – обменник), SLC42A2, SLC42A3 (транспортеры аммиака в каналах RhBG и RhCG) на возможность развития ПКА дистального типа [48-52].
В клинической практике наибольшие трудности возникают при диагностике неполной формы ПКА дистального типа. Связано это с тем, что у данной категории больных достаточно часто отсутствуют основные биохимические признаки ПКА дистального типа: гипоцитратурия, гиперкальциурия, гиперхлоремия [53]. В то же время, несмотря на наличие у данной категории пациентов не столь выраженных метаболических литогенных нарушений, как при полной форме ПКА, процессы камнеобразования протекают достаточно интенсивно.
По настоящему пионерской является работа швейцарских коллег. Viola D’Ambrosio и соавт. изучили наличие генетических мутаций у пациентов с неполной формой ПКА дистального типа. Авторы исследовали биоматериал 22 пациентов с семейным анамнезом МКБ и подтвержденной неполной формой дистального ПКА. В процессе работы были выполнены генетические тестирования с секвенированием 5 генов: SLC4A1 (OMIM 109270), ATP6V1B1 (OMIM 192132), ATP6V0A4 (OMIM 605239), FOXI1 (OMIM 601093) и WDR72 (OMIM 613214) и были обнаружены 2 гетерозиготные мутации в гене SLC4A1 у 2 исследуемых пациентов [14].
Помимо врожденной формы ПКА выделяют также и приобретенную. Одним из основных критериев клинического течения данного типа заболевания является то, что манифестация процессов литогенеза у пациентов встречается во взрослом возрасте и связана с наличием некоторых сопутствующих заболеваний.
Одним из заболеваний, приводящим к ПКА, является синдром Шегрена – аутоиммунное заболевание, характеризующееся поликлональной активацией Вклеток, а также лимфоцитарной инфильтрацией экзокринных желез, приводящей к развитию паренхиматозного сиалоаденита с ксеростомией и сухого кератоконъюнктивита с гиполакримией [54]. При этом совместно с ПКА дистального типа у больных с синдромом Шегрена могут встречаться бессимптомные изменения в анализах мочи, синдром Фанкони, несахарный диабет, гломерулонефрит и тубулоинтерстициальный нефрит [55]. Необходимо подчеркнуть, что синдром Шегрена может быть как первичным, связанным с некоторыми генетическими мутациями, так и вторичным. По данным различных авторов поражение почек при синдроме Шегрена встречается в 4,2–67% случаев [55]. Существенный разброс в цифрах обусловлен, в первую очередь, малочисленными группами исследуемых пациентов, диагностическими критериями и различным дизайном исследований.
Сообщается, что помимо синдрома Шегрена к вторичному ПКА дистального типа могут приводить и другие аутоиммунные заболевания, такие как серонегативная спондилоартропатия, системная красная волчанка, ревматоидный артрит, тиреоидит Хашимото [56-58].
Коллеги из Индии опубликовали работу, в которой сообщается, что ПКА дистального типа у детей может быть связан с пузырно-мочеточниковым рефлюксом (ПМР). При этом ПКА возникает, в основном, у пациентов с запущенными и нелечеными случаями ПМР [59].
Опубликована информация, согласно которой, выполнение сеансов дистанционной литотрипсии может приводить к вторичному ПКА за счет ограниченного повреждения почечных канальцев и возникновения локальных изменений в составе мочи, что теоретически может способствовать росту мочевого камня, состоящего из кальций-фосфата [60, 61].
Достаточно важно подчеркнуть тот факт, что нелеченые и запущенные случаи ПКА могут приводить к поражению опорно-двигательного аппарата – чаще всего у подобных пациентов возникает рахит и остеомаляция. Также могут быть переломы, псевдопереломы, вторичный остеопороз и остеосклероз. Поражение костной ткани связано с метаболическим ацидозом, гиперкальциурией и вторичным гиперпаратиреозом [62, 63].
Сообщается, что причиной ПКА у взрослых может быть прием некоторых лекарственных препаратов. A. Sonnenblick и соавт. опубликовали статью, где утверждают, что прием химиопрепаратов капецитабин, оксалиплатин и цетуксимаб может приводить к ПКА дистальной формы [64].
Опубликованы работы, в которых сообщается, что регулярный прием противоэпилептического средства топирамат, относящегося к классу сульфамат-замещенных моносахаридов, может приводить к ПКА [65].
Сообщается о случаях ПКА дистального типа у пациентов с ВИЧ инфекцией, получающих противовирусную терапию препаратом тенофовира. При этом исследователи отмечают, что после отмены препарата состояние пациентов улучшалось и метаболические показатели приходили в норму [66, 67].
Представлены работы, в которых указано, что прием иммуносупрессивных препаратов, относящихся к группе ингибиторов кальциневрина (сиролимус, такролимус), в 17% случаев способен приводить к ПКА дистального типа [68].
Регулярный прием препарата ибупрофен также способен вызывать нарушения, проводящие к ПКА [69].
В мировой литературе описано большое количество причин возникновения ПКА дистального типа у пациентов. При этом большинство больных имеют характерные специфические изменения показателей крови и мочи, которые позволяют специалисту заподозрить данный вид тубулопатии у больного.
Одними из основных диагностических методов, позволяющих диагностировать дистальную форму ПКА у пациентов с МКБ, являются: общий анализ мочи, биохимический анализ крови и суточной мочи, а также изучение химического состава мочевых камней (табл. 2).
Таблица 2. Особенности лабораторных показателей крови и мочи, а также типы мочевых камней у пациентов с МКБ и ПКА дистального типа
Table 2. Characteristics in laboratory parameters of blood and urine, as well as types of urinary stones in patients with urolithiasis and distal RTA
| ПКА дистального типа / RTA distal type | Норма / Norm | |
|---|---|---|
| Общий анализ мочи / General urine analysis | ||
| pH мочи / urine pH | всегда >5,8 always >5.8 |
|
| Биохимический анализ крови / Blood chemistry | ||
| Калий / Potassium | <3,5 ммоль/л / mmol/l | 3,5-5,1 ммоль/л / mmol/l |
| Хлор / Chlorine | >107 ммоль/л / mmol/l | 98-107 ммоль/л / mmol/l |
| Кислотно-щелочной состав крови / Acid-base composition of blood | ||
| pH крови / blood pH | <7,32 | 7,32-7,42 |
| Бикарбонаты крови / Blood bicarbonates | <15,0 или >26,0 ммоль/л <15.0 or >26.0 mmol/L |
21,0-26,0 ммоль/л / mmol/l |
| Биохимический анализ суточной мочи / Biochemical analysis of daily urine | ||
| Кальций / Calcium | >5,0 ммоль/сут / mmol/day | 2,5-5,0 ммоль/сут / mmol/day |
| Цитраты / citrates | <1,67 ммоль/сут / mmol/day | 1,67-6,45 ммоль/сут / mmol/day |
| Химический состав камня The chemical composition of the stone |
Карбонатапатит, брушит Carbonatapatite, brushite |
|
Достаточно важным в клинической практике является оценка показателей pH мочи в динамике. Для пациентов с ПКА характерны показатели pH мочи >5,8 (табл. 2, рис. 3). Таким образом, высокие цифры pH мочи служат первичным диагностическим маркером ПКА, а оценка результатов общего анализа мочи позволяет заподозрить ПКА уже на этапе первичной диагностики [2]. По собственному клиническому опыту можем сказать, что обычно у пациентов с ПКА дистального типа pH мочи стойко ≥6,0.
Установлено, что у пациентов с ПКА, по данным биохимического анализа крови и суточной мочи, основными факторами риска, стимулирующими процессы камнеобразования, являются гиперкальциурия и гипоцитратурия (табл. 2, рис. 3) [18, 19]. В то же время у многих пациентов по данным биохимического анализа крови диагностируется гиперхлоремия и гипокалиемия [2, 7]. При этом необходимо подчеркнуть, что при изучении результатов анализов у пациента с ПКА не всегда встречаются измененными все вышеперечисленные показатели, достаточно часто могут быть ограниченные отклонения.
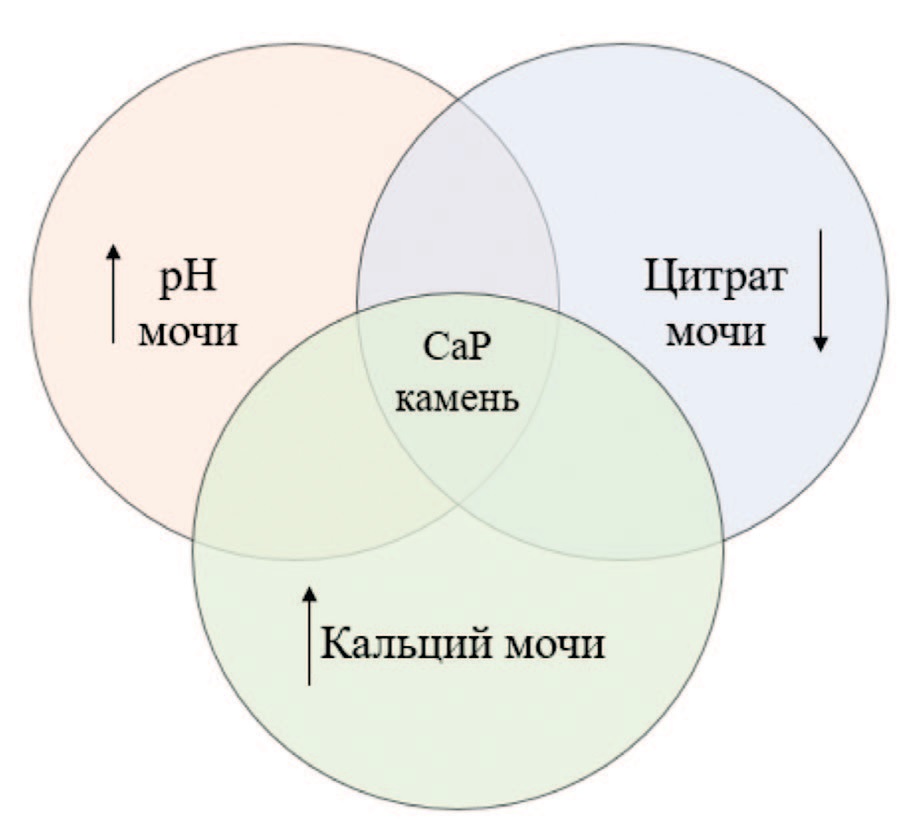
Рис. 3. Основные ключевые литогенные нарушения у пациентов с ПКА дистального типа и МКБ
Fig. 3. The main key lithogenic disorders in patients with distal RTA and urolithiasis
При подозрении на наличие ПКА у пациента с МКБ достаточно информативным является анализ кислотно-щелочного состава крови. Для многих больных с дистальной формой ПКА характерны изменения уровня бикарбонатов крови и низкие показатели pH крови (pH 26,0 ммоль/л) [71].
Согласно данным мировой литературы у пациентов с ПКА и МКБ при химическом анализе мочевых конкрементов могут встречаться как кальций-фосфатные, так и кальций-оксалатные, струвитные и смешанные мочевые камни [23]. В то же время, чаще всего определяются именно кальций-фосфатные камни (брушит или карбонатапатит) [72].
Золотым стандартом диагностики ПКА дистального типа у пациентов с МКБ является применение теста с аммония хлоридом [2].
Алгоритм проведения теста следующий: после пробуждения до 8 часов утра пациент не употребляет пищу. Во время завтрака в 08:00 пациент принимает хлорид аммония, из расчета 0,1 г/кг массы тела. В дальнейшем каждый час проводится сбор мочи и измерение ее кислотности при помощи индикаторных полосок или pH-метра в течение 5 часов после приема препарата. Все результаты регистрируются в бланке протокола теста [73].
Противопоказанием к проведению пробы являются заболевания печени, инфекция мочевых путей, вызванная уреазопродуцирующей микрофлорой, выраженный метаболический ацидоз. Результаты теста трактуются следующим образом:
• при рН мочи <5,4 в одном из пяти замеров мочи у пациента исключается ПКА;
• при рН 5,5 – 6,0 во всех пяти замерах – диагностируется неполный ПКА;
• при рН мочи ≥ 6 во всех пяти замерах – диагностируется полный ПКА [73].
Несмотря на наличие апробированного и одобренного метода диагностики ПКА дистального типа, возникают определенные трудности с выявлением ПКА. Данный факт связан с тем, что в аптечных сетях практически невозможно приобрести аммония хлорид.
Европейские коллеги предложили иной подход к диагностике ПКА с использованием фуросемида и флудрокортизована в качестве альтернативы аммоний хлориду [74, 75]. Считается, что данный метод можно использовать для выявления ПКА, при этом диагностическая проба выполняется по абсолютно идентичной схеме, принятой для аммония хлорида. Несмотря на наличие положительных заключений о возможности применения пробы с фуросемидом и флудрокортизованом в качестве метода диагностики ПКА, пока она остается во многом экспериментальной, требуется проведение дополнительных научных исследований по данной тематике.
Пациенты с диагнозом МКБ и ПКА дистального типа должны регулярно наблюдаться у уролога и нефролога, контролировать основные биохимические показатели крови и суточной мочи и постоянно получать противорецидивную терапию.
Регулярное применение цитратных смесей (соли цитрата, в основном, калия цитрат) является стандартом метафилактики МКБ на фоне дистальной формы ПКА [76-78]. На российском фармакологическом рынке в настоящее время присутствуют препараты, содержащие калия гидрокарбонат, лимонную кислоту, натрия цитрат или калия натрия гидроцитрат.
Считаем важным обратить внимание коллег на тот факт, что дозировка цитратных смесей при ПКА дистального типа должна подбираться индивидуально. В некоторых случаях (например, у детей) необходим прием препаратов не только в дневное время, но и в ночные часы [79]. Основным критерием эффективности применения цитратных смесей у данной категории больных является нормализация уровня цитратов в суточной моче и нормализация концентрации бикарбонатов в крови [80].
Рекомендуя пациентам с ПКА терапию цитратными смесями, крайне важно помнить, что назначение препаратов, содержащих калий, у пациентов с гиперкалиемией противопоказано [81].
В некоторых случаях существующие подходы к лечению являются низкоэффективными и могут сопровождаться появлением осложнений со стороны желудочно-кишечного тракта (гастрит, язвенная болезнь желудка, двенадцатиперстной кишки) [82]. Многократный прием препаратов является неудобным для пациентов и снижает приверженность к лечению. Для уменьшения количества осложнений и повышения уровня приверженности больных к терапии в настоящее время разрабатываются пероральные гранулированные препараты пролонгированного действия на основе цитратных смесей (ADV7103) [83].
Наравне с коррекцией гипоцитратурии нормализация уровня кальция в суточной моче является одним из ключевых звеньев в метафилактике МКБ у больных с ПКА дистального типа [2]. Основным лекарственным препаратом для коррекции уровня кальция в моче является гидрохлоротиазид – препарат, относящийся к группе тиазидных диуретиков. Доказано, что гидрохлоротиазид снижает показатели экскреции кальция с мочой [84]. При этом механизмы действия препарата остаются до конца не изученными [3].
Помимо лекарственной терапии пациентам с МКБ и ПКА дистального типа необходимо придерживаться определенных диетических рекомендаций. Основным правилом при соблюдении диеты является потребление большого количества жидкости, в том числе апельсинового свежевыжатого сока или воды с лимоном [85]. Также больным рекомендуется ограничить соль и соленые продукты и снизить количество потребляемого мяса.
ЗАКЛЮЧЕНИЕ
Практикующему урологу важно помнить, что ПКА дистального типа может быть одной из причин литогенеза у пациентов с рецидивирующим течением МКБ. Основными диагностическими критериями являются специфические изменения показателей биохимического анализа крови, суточной мочи, а также особые показатели pH мочи. При этом у некоторых пациентов с ПКА и МКБ могут отсутствовать характерные изменения. Проведение специализированной пробы помогает своевременно диагностировать ПКА и назначить адекватную противорецидивную терапию.
ЛИТЕРАТУРА
1. Мартов А.Г., Харчилава Р.Р., Акопян Г.Н., Гаджиев Н.К., Мазуренко Д.А., Малхасян В.А. Мочекаменная болезнь. Клинические рекомендации 2020;53 с. URL: http://disuria.ru/_ld/7/733_kr20N20mz.pdf. [Martov A.G., Kharchilava R.R., Akopyan G.N., Gadzhiev N.K., Mazurenko D.A., Malkhasyan V.A. Urolithiasis disease. Clinical guidelines 2020;53 p. URL: http://disuria.ru/_ld/7/733_kr20N20mz.pdf. (In Russian)].
2. Türk C, Skolarikos A, Neisius A, Petřík A, Seitz C, Thomas K. Guidelines on Urolithiasis 2021 European Urology Association. URL: www.uroweb.org.
3. Coe FL, Worcester EM, Evan AP. Idiopathic hypercalciuria and formation of calcium renal stones. Nat Rev Nephrol 2016;12(9):519-33. https://doi.org/10.1038/nrneph.2016.101.
4. Lewandowski S, Rodgers AL. Idiopathic calcium oxalate urolithiasis: risk factors and conservative treatment. Clin Chim Acta 2004;345(1-2):17-34. https://doi.org/10.1016/j.cccn.2004.03.009.
5. Alaya A, Sakly R, Nouri A, Najjar MF, Belgith M, Jouini R. Idiopathic urolithiasis in Tunisian children: a report of 134 cases. Saudi J Kidney Dis Transpl 2013;24(5):1055-61. https://doi.org/10.4103/1319-2442.118099.
6. Bu Q, Zhu Y, Chen QY, Li H, Pan Y. A polymorphism in the 3'-untranslated region of the matrix metallopeptidase 9 gene is associated with susceptibility to idiopathic calcium nephrolithiasis in the Chinese population. J Int Med Res 2020;48(12):300060520980211. https://doi.org/10.1177/0300060520980211.
7. Козыро И.А., Сукало А.В., Белькевич А.Г. Тубулопатии у детей: учебно-методическое пособие. Минск: БГМУ 2019; 26 с. [Kozyro I.A., Sukalo A.V., Belkevich A.G. Tubulopathy in children: educational and methodical manual. Minsk: BSMU 2019; 26 p. (In Russian)].
8. Juan Rodríguez Soriano. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol 2002;13(8):2160-70. https://doi.org/10.1097/01.asn.0000023430.92674.e5.
9. Савин И.А., Горячев А.С. Водно-электролитные нарушения в нейрореанимации. М., Медицинские книги 2016; 115-119 с. [Savin I.A., Goryachev A.S. Water-electrolyte disturbances in neuroreanimation. M., Medical Books 2016; 115-119 p. (In Russian)].
10. Полуднякова Л.В., Абакумова Т.В., Долгова Д.Р., Генинг Т.П., Михайлова Н.Л. Физиология выделения: учеб. пособие к практическим занятиям по нормальной физиологии человека для студентов медицинского факультета. Ульяновск: УлГУ 2018; 28 с. [Poludnyakova L.V., Abakumova T.V., Dolgova D.R., Gening T.P., Mikhailova N.L. Physiology of excretion: textbook. manual for practical exercises on normal human physiology for students of the medical faculty. Ulyanovsk: UlGU 2018; 28 p. (In Russian)].
11. Mohebbi N, Wagner CA. Pathophysiology, diagnosis and treatment of inherited distal renal tubular acidosis. J Nephrol 2018;31(4):511-522. https://doi.org/10.1007/s40620-017-0447-1.9.
12. Михайленко Б.Ю. Основные механизмы регуляция гидрокарбонатной буферной системы в рамках поддержания гомеостаза кислотно-щелочного равновесия организма человека. The Scientific Heritage 2021;68(2):14-16. [Mikhailenko B. The main mechanisms of regulation of the bicarbonate buffer system in the framework of maintaining homeostasis of acid-base balance of the human body. The Scientific Heritage 2021;68(2):14-16. (In Russian)]. https://doi.org/10.24412/9215-0365-2021-68-2-14-16.
13. Рахматуллина А.С., Дехтярь Т.Ф. Роль буферных систем в организме человека. Современные научные исследования и разработки 2018;1(12(29)):528-530. [Rakhmatullina A.S., Dekhtyar T.F. Buffer systems in the human body. Sovremennyye nauchnyye issledovaniya i razrabotki = Modern scientific researches and innovations 2018;1(12(29)):528-530. (In Russian)].
14. D'Ambrosio V, Azzarà A, Sangiorgi E, Gurrieri F, Hess B, Gambaro G, Ferraro PM. Results of a gene panel approach in a cohort of patients with incomplete distal renal tubular acidosis and nephrolithiasis. Kidney Blood Press Res 2021;46(4):469-474. https://doi.org/10.1159/000516389.
15. Santos F, Gil-Peña H, Alvarez-Alvarez S. Renal tubular acidosis. Curr Opin Pediat 2017;29(2):206-210. https://doi.org/10.1097/MOP.0000000000000460.
16. Детская нефрология: практическое руководство. Под ред. Э. Лоймана, А. Н. Цыгина, А. А. Саркисяна. М.: ЛитТерра 2010; 400 с. Pediatric nephrology: a practical guide. ed. E. Loiman, A. N. Tsygin, A. A. Sarkisyan. M.: LitTerra 2010; 400 p. (In Russian)].
17. Brenner RJ, Spring DB, Sebastian A, McSherry EM, Genant HK, Palubinskas AJ, et al. Incidence of radiographically evident bone disease, nephrocalcinosis, and nephrolithiasis in various types of renal tubular acidosis. New Engl J Med 1982;307(4):217-21. https://doi.org/10.1056/NEJM198207223070403.
18. Caruana RJ, Buckalew VM Jr. The syndrome of distal (type I) renal tubular acidosis. Clinical and laboratory findings in 58 cases. Medicine 1988;67(2):84-99. https://doi.org/10.1097/00005792-198803000-00002.
19. Buckalew VM Jr, Caruana RJ. The pathophysiology of distal (type 1) renal tubular acidosis. In: Gonick H.C., Buckalew V.M. Renal Tubular Disorders: Pathophysiology, Diagnosis and Management 1985; New York: Marcel Dekker; 357-386 p.
20. Wrong O, Davies HE. The excretion of acid in renal disease. Q J Med 1959;28(110):259–313
21. Batlle D, Moorthi KM, Schlueter W, Kurtzman N. Distal renal tubular acidosis and the potassium enigma. Semin Nephrol 2006(26):471–478. https://doi.org/10.1016/j.semnephrol.2006.12.001.
22. Arampatzis S, Ropke-Rieben B, Lippuner K, Hess B. Prevalence and densitometric characteristics of incomplete distal renal tubular acidosis in men with recurrent calcium nephrolithiasis. Urol Res 2012;40(1):53–59. https://doi.org/10.1007/s00240-011-0397-3.
23. Buckalew VM Jr. Nephrolithiasis in renal tubular acidosis. J Urol 1989;141(3 Pt 2):731-7. https://doi.org/10.1016/s0022-5347(17)40997-9.
24. Magni G, Unwin RJ, Moochhala SH. Renal tubular acidosis (RTA) and kidney stones: diagnosis and management. Arch Esp Urol 2021;74(1):123-128.
25. He BJ, Liao L, Deng ZF, Tao YF, Xu YC, Lin FQ. Molecular genetic mechanisms of hereditary spherocytosis: current perspectives. Acta Haematol 2018;139(1):60-66. https://doi.org/10.1159/000486229.
26. Стяжкина С.Н., Маннанова Д.Р., Галяутдинова А.И. Течение микросфероцитоза Минковсвого-Шоффара на фоне спленомегалии. Клинический случай. Modern Science 2020;10(2):317-321. [Styazhkina S.N., Mannanova D.R., Galyautdinova A.I. The course of microspherocytosis of Minkowsvogo-Choffard against the background of splenomegaly. Clinical case. Modern Science 2020;10(2):317-321. (in Russian)].
27. Михеева О.М., Акопова А.О., Охапкина Т.Г., Ивкина Т.И. Гастроэнтерологические осложнения у больных с наследственным микросфероцитозом. Медицинский алфавит 2015;1(7): 44-46. [Mikheeva O.M., Akopova A.O., Okhapkina T.G., Ivkina T. Gastrointestinal complications in patients with hereditary microspherocytosis. Meditsinskiy alfavit = Medical Alphabet 2015;1(7):44-46. (In Russian)].
28. Wagner CA, Imenez Silva PH, Bourgeois S. Molecular pathophysiology of acid-base disorders. Semin Nephrol 2019;39(4):340-352. https://doi.org/10.1016/j.semnephrol.2019.04.004.
29. Soares SBM, de Menezes Silva LAW, de Carvalho Mrad FC, Simões E Silva AC. Distal renal tubular acidosis: genetic causes and management. World J Pediatr 2019;15(5):422-431. https://doi.org/10.1007/s12519-019-00260-4.
30. Ito N, Ihara K, Kamoda T, Akamine S, Kamezaki K, Tsuru N, et al. Autosomal dominant distal renal tubular acidosis caused by a mutation in the anion exchanger 1 gene in a Japanese family. CEN Case Rep 2015(4):218–22.
31. Chu C, Woods N, Sawasdee N, Guizouarn H, Pellissier B, Borgese F, et al. Band 3 Edmonton I, a novel mutant of the anion exchanger 1 causing spherocytosis and distal renal tubular acidosis. Biochem J 2010(426):379–388. https://doi.org/10.1042/BJ20091525.
32. Khositseth S, Bruce LJ, Walsh SB, Bawazir WM, Ogle GD, Unwin RJ, et al. Tropical distal renal tubular acidosis: clinical and epidemiological studies in 78 patients. QJM 2012;105(9):861- 77. https://doi.org/10.1093/qjmed/hcs139.
33. Кирпичникова А.А., Чэнь Т., Романюк Д.А., Емельянов В.В., Шишова М.Ф. Особенности регуляции вакуолярной H+-АТФазы растительных клеток. Вестник Санкт-Петербургского университета Серия 3 «Биология» 2016;3(2):149-60. [Kirpichnikova А.А., Chen Т., Romanyuk D.А., Yemelyanov V.V., Shishova M.F. Peculiar features of plant cell vacuolar h+-atpase regulation. Vestnik Sankt-Peterburgskogo universiteta Seriya 3 Biologiya = Vestnik of Saint Petersburg University. Series 3. Biology 2016;3(2):149-60. (In Russian)]. https://doi.org/10.21638/11701/spbu03.2016.212.
34. Dettmer J, Liu TY, Schumacher K. Functional analysis of Arabidopsis V-ATPase subunit VHA-E isoforms. Eur J Cell Biol 2010(89):152‒156. https://doi.org/10.1016/j.ejcb.2009.11.008.
35. Mauvezin C, Nagy P, GaborJuha SZ, Neufeld TP. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidifi cation. Nat Commun 2015(6):7007. https://doi.org/10.1038/ ncomms8007.
36. Wagner CA, Finberg KE, Breton S, Marshansky V, Brown D, Geibel JP. Renal vacuolar H+- ATPase. Physiol Rev 2004t;84(4):1263-314. https://doi.org/10.1152/physrev.00045.2003.
37. Kawasaki-Nishi S, Nishi T, Forgac M. Proton translocation driven by ATP hydrolysis in VATPases. FEBS Lett 2003;545(1):76–85. https://doi.org/10.1016/s0014-5793(03)00396-x.
38. Adadey SM, Wonkam-Tingang E, Aboagye ET, Quaye O, Awandare GA, Wonkam A. Hearing loss in Africa: current genetic profile. Hum Genet 2022;141(3-4):505-517 https://doi.org/10.1007/s00439-021-02376-y.
39. Ruf R, Rensing C, Topaloglu R, Guay-Woodford L, Klein C, Volmer M, et al. Confrmation of the ATP6B1 gene as responsible for distal renal tubular acidosis. Pediatr Nephrol 2003(18):105–9. https://doi.org/10.1007/s00467-002-1018-8.
40. Pathare G, Dhayat N, Mohebbi N, Wagner CA, Bobulescu IA, Moe OW, et al. Changes in V-ATPase subunits of human urinary exosomes refect the renal response to acute acid/alkali loading and the defects in distal renal tubular acidosis. Kidney Int 2018(94):871–80. https://doi.org/10.1016/j.kint.2017.10.018.
41. Karet FE, Finberg KE, Nayir A, Bakkaloglu A, Ozen S, Hulton SA, et al. Localization of a gene for autosomal recessive distal renal tubular acidosis with normal hearing (rdRTA2) to 7q33-34. Am J Hum Genet 1999;65(6):1656-65. https://doi.org/10.1086/302679.
42. Enerback S, Nilsson D, Edwards N, Heglind M, Alkanderi S, Ashton E, et al. Acidosis and deafness in patients with recessive mutations in FOXI1. J Am Soc Nephrol 2018;29(3):1041-8. https://doi.org/10.1681/ASN.2017080840.
43. Vidarsson H, Westergren R, Heglind M, Blomqvist SR, Breton S, Enerback S. The forkhead transcription factor Foxi1 is a master regulator of vacuolar H-ATPase proton pump subunits in the inner ear, kidney and epididymis. PLoS One 2009;4(2):e4471. https://doi.org/10.1371/journal.pone.0004471. Epub 2009 Feb 13.
44. Blomqvist SR, Vidarsson H, Fitzgerald S, Johansson BR, Ollerstam A, Brown R, et al. Distal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1. J Clin Invest 2004;113(11):1560-70. https://doi.org/10.1172/JCI20665.
45. Blomqvist SR, Vidarsson H, Soder O, Enerback S. Epididymal expression of the forkhead transcription factor Foxi1 is required for male fertility. EMBO J 2006;25(17):4131-41. https://doi.org/10.1038/sj.emboj.7601272.
46. Benonisdottir S, Kristjansson RP, Oddsson A, Steinthorsdottir V, Mikaelsdottir E, Kehr B, et al. Sequence variants associating with urinary biomarkers. Hum Mol Genet 2019;28(7):1199-211. https://doi.org/10.1093/hmg/ddy409.
47. Rungroj N, Nettuwakul C, Sawasdee N, Sangnual S, Deejai N, Misgar RA, et al. Distal renal tubular acidosis caused by tryptophan-aspartate repeat domain 72 (WDR72) mutations. Clin Genet 2018;94(5):409-18. https://doi.org/10.1111/cge.13418.
48. Boettger T, Hubner CA, Maier H, Rust MB, Beck FX, Jentsch TJ. Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature 2002(416):874-8. https://doi.org/10.1038/416874a.
49. Xu J, Song P, Nakamura S, Miller M, Barone S, Alper SL, et al. Deletion of the chloride transporter slc26a7 causes distal renal tubular acidosis and impairs gastric acid secretion. J Biol Chem 2009;284(43):29470-9. https://doi.org/10.1074/jbc.M109.044396.
50. Gao X, Eladari D, Leviel F, Tew BY, Miró-Julià C, Cheema FH, et al. Deletion of hensin/DMBT1 blocks conversion of {beta}- to {alpha}-intercalated cells and induces distal renal tubular acidosis. Proc Natl Acad Sci USA 2010;107(50):21872-7. https://doi.org/10.1073/ pnas.1010364107.
51. Schwartz GJ, Gao X, Tsuruoka S, Purkerson JM, Peng H, D'Agati V, et al. SDF1 induction by acidosis from principal cells regulates intercalated cell subtype distribution. J Clin Invest 2015;125(12):4365-74. https://doi.org/10.1172/JCI80225.
52. Biver S, Belge H, Bourgeois S, Van Vooren P, Nowik M, Scohy S, et al. A role for Rhesus factor Rhcg in renal ammonium excretion and male fertility. Nature 2008;456(7220):339-43. https://doi.org/10.1038/nature07518.
53. Fuster DG, Moe OW. Incomplete Distal Renal Tubular Acidosis and Kidney Stones. Adv Chronic Kidney Dis 2018;25(4):366-374. https://doi.org/10.1053/j.ackd.2018.05.007.
54. Головач И.Ю., Егудина Е.Д. Особенности поражения почек при системных заболеваниях соединительной ткани. Почки 2018;7(4):275-90. [Golovach I.Yu., Yehudina Ye.D. Features of renal involvement in systemic connective tissue diseases. Pochki = Kidneys 2018;7(4):275-90. (In Russian)]. https://doi.org/10.22141/2307-1257.7.4.2018.148517.
55. François H, Mariette X. Renal involvement in primary Sjögren syndrome. Nat Rev Nephrol 2016;12(2):82-93. https://doi.org/10.1038/nrneph.2015.174.
56. Agrawal N, Mahata R, Chakraborty PP, Basu K. Secondary distal renal tubular acidosis and sclerotic metabolic bone disease in seronegative spondyloarthropathy. BMJ Case Rep 2022;15(3):e248712. https://doi.org/10.1136/bcr-2021-248712.
57. Giglio S, Montini G, Trepiccione F, Gambaro G, Emma F. Distal renal tubular acidosis: a systematic approach from diagnosis to treatment. J Nephrol 2021;34(6):2073-2083. https://doi.org/10.1007/s40620-021-01032-y.
58. Wang S, Dong B, Wang C, Lu J, Shao L. From Bartter's syndrome to renal tubular acidosis in a patient with Hashimoto's thyroiditis: A case report. Clin Nephrol 2020;94(3):150-54. https://doi.org/10.5414/CN109820.
59. Bharani A, Bharani T, Bharani R. Distal renal tubular acidosis secondary to vesico-ureteric reflux: A case report with review of literature. Saudi J Kidney Dis Transpl 2018;29(5):1240-1244. https://doi.org/10.4103/1319-2442.243943.
60. Evan AP, Willis LR, Lingeman JE, McAteer JA. Renal trauma and the risk of long-term complications in shock wave lithotripsy. Nephron 1998;78(1):1–8. https://doi.org/10.1159/000044874.
61. Parks JH, Worcester EM, Coe FL, Evan AP, Lingeman JE. Clinical implications of abundant calcium phosphate in routinely analyzed kidney stones. Kidney Int 2004;66(2):777–85. https://doi. org/10.1111/j.1523-1755.2004.00803.x.
62. Boro H, Khatiwada S, Alam S, Kubihal S, Dogra V, Mannar V, et al. Renal Tubular Acidosis Manifesting as Severe Metabolic Bone Disease. touchREV Endocrinol 2021;17(1):59-67. https://doi.org/10.17925/EE.2021.17.1.59.
63. Rajan R, Cherian KE, Mathew J, Asha HS, Kapoor N, Paul TV. Beyond sicca symptoms: Osteomalacia secondary to renal tubular acidosis in Sjogren syndrome. Joint Bone Spine 2021;88(1):105064. https://doi.org/10.1016/j.jbspin.2020.07.013.
64. Sonnenblick A, Meirovitz A. Renal tubular acidosis secondary to capecitabine, oxaliplatin, and cetuximab treatment in a patient with metastatic colon carcinoma: a case report and review of the literature. Int J Clin Oncol 2010;15(4):420-2. https://doi.org/10.1007/s10147-010-0050-0.
65. Izzedine H, Launay-Vacher V, Deray G. Topiramate-induced renal tubular acidosis. Am J Med 2004;116(4):281-2. https://doi.org/10.1016/j.amjmed.2003.08.021.
66. Salimi R, Begum I, Varma DM, Nandakrishna B, Rajesh R, Vidyasagar S. Tenofovir disoproxil fumarate-induced distal renal tubular acidosis: A case report. Int J STD AIDS 2020;31(3):276-279. https://doi.org/10.1177/0956462419887877.
67. Iwata K, Nagata M, Watanabe S, Nishi S. Distal renal tubular acidosis without renal impairment after use of tenofovir: a case report. BMC Pharmacol Toxicol 2016;17(1):52. https://doi.org/10.1186/s40360-016-0100-y.
68. Banhara PB, Gonçalves RT, Rocha PT, Delgado AG, Leite M Jr, Gomes CP. Tubular dysfunction in renal transplant patients using sirolimus or tacrolimus. Clin Nephrol 2015 Jun;83(6):331-7. https://doi.org/10.5414/CN108541.
69. Li R, Hasan N, Armstrong L, Cockings J. Impaired consciousness, hypokalaemia and renal tubular acidosis in sustained Nurofen Plus abuse. BMJ Case Rep 20197;12(11):e231403. https://doi.org/10.1136/bcr-2019-231403.
70. Баранов А.А., Намазова-Баранова Л.С., Цыгин А.Н., Сергеева Т.В., Чумакова О.В., Паунова С.С., и др. Тубулопатии у детей. Клинические рекомендации 2016; 57 с. [Baranov A.A., Namazova-Baranova L.S., Tsygin A.N., Sergeeva T.V., Chumakova O.V., Paunova S.S., et al. Tubulopathies in children. Clinical guidelines 2016;57 p. (In Russian)]. URL: https://www.pediatr-russia.ru/information/klin-rek/deystvuyushchie-klinicheskie rekomendatsii/%D0%A2%D1%83%D0%B1%D1%83%D0%BB%D0%BE %D0%BF%D0%B0%D1%82% D0%B8%D0%B8%20%D0%A1%D0%9F%D0%A0.v1%2028%2002%202017-1.pdf.
71. Palmer BF, Kelepouris E, Clegg DJ. Renal tubular acidosis and management strategies: a narrative review. Adv Ther 2021;38(2):949-968. https://doi.org/10.1007/s12325-020-01587-5.
72. Daudon M, Bouzidi H, Bazin D. Composition and morphology of phosphate stones and their relation with etiology. Urol Res 2010(38):459–467.
73. Голованов С.А., Сивков А.В., Анохин Н.В. Гиперкальциурия: принципы дифференциальной диагностики. Экспериментальная и клиническая урология 2015;(4):86-92. [Golovanov S.A., Sivkov A.V., Anokhin N.V. Hypercalciuria: principles of differential diagnostics. Eksperimentalnaya i Klinicheskaya urologiya = Experimental and Clinical Urology 2015;(4):86-92. (In Russian)].
74. Dhayat NA, Gradwell MW, Pathare G, Anderegg M, Schneider L, Luethi D, et al. Furosemide/Fludrocortisone test and clinical parameters to diagnose incomplete distal renal tubular acidosis in kidney stone formers. Clin J Am Soc Nephrol 2017;12(9):1507-1517. https://doi.org/10.2215/CJN.01320217.
75. Walsh SB, Shirley DG, Wrong OM, Unwin RJ. Urinary acidification assessed by simultaneous furosemide and fludrocortisone treatment: an alternative to ammonium chloride. Kidney Int 2007;71(12):1310-6. https://doi.org/10.1038/sj.ki.5002220.
76. Soriano JR. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol 2002;13(8):2160–2170. https://doi.org/10.1097/01.asn.0000023430.92674.e5.
77. Alonso-Varela M, Gil-Pena H, Coto E, Gómez J, Rodríguez J, Rodríguez-Rubio E, et al. Distal renal tubular acidosis. Clinical manifestations in patients with different underlying gene mutations. Pediatr Nephrol 2018;33(9):1523–1529. https://doi.org/10.1007/s00467-018-3965-8.
78. Both T, Zietse R, Hoorn EJ, van Hagen PM, Dalm VA, van Laar JA, et al. Everything you need to know about distal renal tubular acidosis in autoimmune disease. Rheumatol Int 2014;34(8):1037–1045. https://doi.org/10.1007/s00296-014-2993-3.
79. Chan JCM, Santos F. Renal tubular acidosis in childhood. World J Pediatr 2007(3):92–97.
80. Watanabe T. Improving outcomes for patients with distal renal tubular acidosis: recent advances and challenges ahead. Pediatric Health Med Ther 2018(9):181–90. https://doi.org/10.2147/PHMT.S174459.
81. Bagga A, Sinha A. Renal Tubular Acidosis. Indian J Pediatr 2020;87(9):733-744. https://doi.org/10.1007/s12098-020-03318-8. Epub 2020 Jun 26.
82. Lopez-Garcia SC, Emma F, Walsh SB, Fila M, Hooman N, Zaniew M, et al. Treatment and long-term outcome in primary distal renal tubular acidosis. Nephrol Dial Transplant 2019;34(6):981–991. https://doi.org/10.1093/ndt/gfy409.
83. Bertholet-Thomas A, Guittet C, Manso-Silván MA, Castang A, Baudouin V, Cailliez M, et al. Efficacy and safety of an innovative prolonged-release combination drug in patients with distal renal tubular acidosis: an open-label comparative trial versus standard of care treatments. Pediatr Nephrol 2021;36(1):83-91. https://doi.org/10.1007/s00467-020-04693-2.
84. Fink HA, Wilt TJ, Eidman KE, Garimella PS, MacDonald R, Rutks IR, Brasure M, et al. Medical management to prevent recurrent nephrolithiasis in adults: a systematic review for an American College of Physicians Clinical Guideline. Ann Intern Med 2013;158(7):535–543. https://doi.org/10.7326/0003-4819-158-7-201304020-00005.
85. Ferraro PM, Taylor EN, Gambaro G, Curhan GC. Soda and other beverages and the risk of kidney stones. Clin J Am Soc Nephrol 2013;8(8):1389-95. https://doi.org/10.2215/CJN.11661112.
